Computational Biology
A Brief History of Virtual Cells
The idea of computationally modeling a cell is not new. What's new is that it might actually work.
The field has gone through two distinct eras — each built on a different theory of how to understand a cell, each running into the same fundamental wall, and each leaving something the next era needed. Here's what actually happened.
Virtual Cell 1.0: If You Know the Biology, Simulate It

The founding premise of the first era was straightforward: a cell is a biochemical machine. If you wrote down all the reactions — every enzyme, every metabolic flux, every regulatory interaction — you could simulate it. Code it up, run it, and the biology falls out.
The first serious attempt was E-Cell, launched in 1996 by Masaru Tomita's lab at Keio University and published in 1997 (Tomita et al., Genome Informatics, 1997; extended in Bioinformatics, 1999). They modeled a hypothetical minimal organism with 127 genes drawn from Mycoplasma genitalium — simulating transcription, translation, energy production, and phospholipid synthesis as coupled differential equations. It worked, in a narrow sense. You could perturb a simulated gene and watch the downstream effects propagate through metabolism.
But it didn't scale. Biology has millions of reactions, most of them poorly characterized. Every parameter required experimental measurement. Every new organism required a new model built from scratch.
The 2000s produced a more tractable variant: genome-scale metabolic models. Rather than simulating full dynamics, these used constraint-based linear programming to predict which metabolic fluxes were feasible given a cell's known reactions. Bernhard Palsson's group at UCSD led this effort, producing genome-scale reconstructions of E. coli, yeast, and eventually human cell lines. Scientifically useful for studying metabolism and predicting growth under specific conditions — but they captured only the metabolic layer. Signaling, transcription, translation, and cell division remained outside their scope.
The landmark of this era came in 2012. Jonathan Karr and colleagues published the first whole-cell computational model — a complete simulation of Mycoplasma genitalium in Cell (Karr et al., Cell, 2012, 150:389–401). They modeled all 525 genes and 28 biological processes — DNA replication, transcription, translation, metabolism, cell division — as an integrated simulation. It took years to build and required extraordinary biological expertise to parameterize. But it proved the concept: given enough known biochemistry, you could simulate a cell well enough to predict how it behaves.
The problem was immediately obvious. Mycoplasma genitalium has 525 genes. A human cell has roughly 20,000, embedded in signaling and regulatory networks orders of magnitude more complex — most of which we don't understand well enough to write down as equations. Virtual Cell 1.0 hit a wall you can't curate your way past.
Virtual Cell 2.0: If You Have Enough Data, Learn It

What changed everything was data — and a new generation of models built to learn from it.
Starting around 2014, high-throughput single-cell sequencing became practical. Drop-seq (Macosko et al., Cell, May 2015) and inDrop (Klein et al., Cell, May 2015) brought the cost of profiling individual cells down to pennies and made large-scale experiments routine. Rather than measuring gene expression in bulk tissue — an average over millions of mixed cell types — you could now profile individual cells and see the exact transcriptional state of each one. By 2016, when the Human Cell Atlas launched with the goal of cataloguing every cell type in the human body, the field was generating data at a scale nobody had anticipated. The question shifted: instead of "can we manually encode the biology?", it became "can we learn it from data?"
The first real answer came in 2019. scGen (Lotfollahi, Wolf & Theis, Nature Methods, 2019, 16:715–721) used a variational autoencoder to predict how cells respond to perturbations — not by simulating biochemistry, but by learning latent structure from data. Given the transcriptional profile of a cell in a control condition, it could predict what that profile would look like after treatment, generalizing across cell types and species. It worked better than anything before it and pointed the field in a new direction: instead of building models by hand, let the data do the teaching.
Then transformers arrived — and the field moved fast.
The first foundation model for single-cell biology was scGPT (Cui, Wang et al., preprint April 2023; Nature Methods, 2024). Pre-trained on 33 million human cells from CellxGene using a generative transformer architecture, it was designed from the start as a multi-task model: the same pre-trained weights fine-tuned across cell type annotation, perturbation response prediction, batch correction, multi-omic integration, and gene regulatory network inference. The key question scGPT asked — and answered — was whether a single foundation model could generalize across the diverse tasks that understanding a cell actually requires. It could.
Geneformer (Theodoris et al., Nature, May 2023, Vol. 618) followed closely, pre-training on 30 million single-cell profiles and representing each cell as a rank-ordered sequence of genes by expression level. It transferred usefully to predicting gene dosage sensitivity in rare cardiac disease contexts — demonstrating that pre-trained representations could encode genuine regulatory logic.
The ecosystem expanded rapidly from there. scFoundation (Hao et al., preprint May 2023; Nature Methods, 2024) scaled pre-training to 50 million cells with a read-depth-aware architecture designed to handle the heavy dropout noise that plagues single-cell data. UCE (Rosen et al., 2023) aimed for universal embedding — trained across 36 million cells from 150 cell types and multiple species, targeting zero-shot transfer to cell types never seen during training. Both pushed on the same question: can models that have seen enough biology extrapolate to biology they haven't?
GEARS (Roohani, Huang & Leskovec, Nature Biotechnology, August 2023) tackled the hardest downstream task directly: predicting transcriptional outcomes of novel multi-gene perturbations. By combining a graph neural network over known gene interaction networks with a perturbation encoder, it generalized to combinatorial knockouts it hadn't seen — a step toward the kind of reasoning drug discovery actually requires.
By 2024, Virtual Cell 2.0 had become a strategic bet. The Chan Zuckerberg Initiative announced a dedicated program. Xaira Therapeutics launched with a core thesis around building the causally-rich, contextually-diverse perturbation datasets needed to make virtual cells work in drug discovery. Recursion, Isomorphic Labs, and others made similar bets.
What's Still Hard
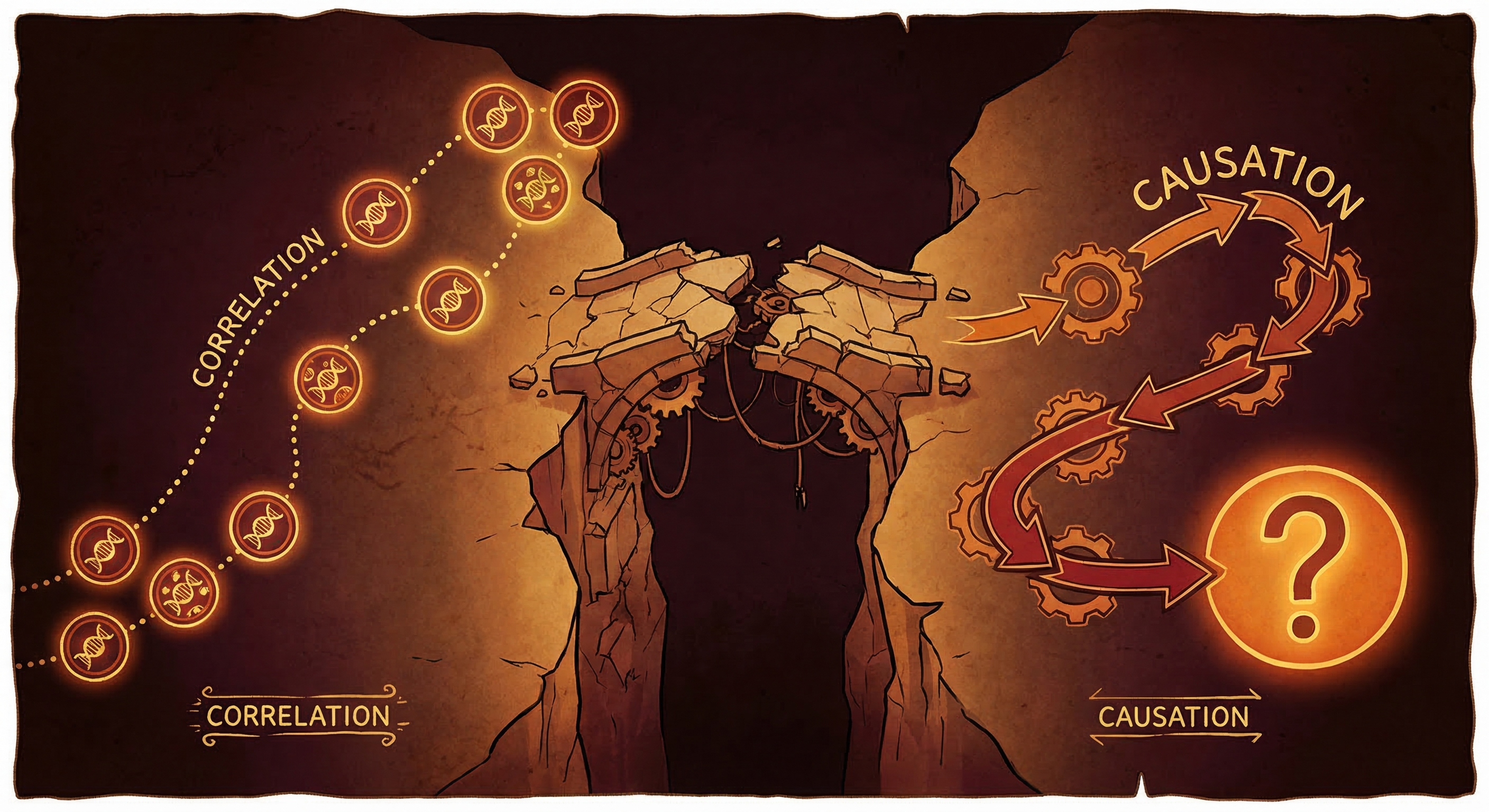
Virtual Cell 2.0 is not Virtual Cell 1.0's problems solved. It's a different set of problems.
The fundamental constraint isn't architecture — it's data. Most public single-cell datasets come from a handful of commonly studied tissues, measured with a handful of commonly used protocols. Models trained on this distribution learn what's common in that distribution. Ask them to generalize to rare cell types, understudied diseases, or novel perturbation classes, and performance drops.
More deeply: current models learn correlations, not causes. A model can learn that drug A tends to downregulate gene X in this cell type. It cannot reliably tell you whether downregulating gene X would be therapeutic or toxic in a new context where the upstream regulators are different. The perturbation problem isn't a retrieval problem. It's a causal reasoning problem — and we're not there yet.
The path forward runs through data diversity first. Not more cells from the same tissues — different tissues, different diseases, different organisms, different perturbation types, different measurement modalities. Perturbation screens at scale, designed specifically to capture the regulatory wiring of the cell rather than just document what happens in convenient conditions.
Two decades after E-Cell, the field has the data, the models, and the compute. What it's still working out is whether the models are actually learning the right things.